



Education
- B.A.: Harvard University, Physics, Sept. 1961 – June 1965
- Ph.D.: University of Chicago, Chemical Physics, Sept. 1965 – Sept. 1969
- Post-Doctoral Research:
- i.) University of California, San Diego-Chemical Physics, Sept. 1969 – Sept. 1971
- ii) Cambridge University-Physics, Sept. 1971 – Sept. 1972
Professional Experience
- 1995-present: Distinguished University Professor, University of Maryland
- 1990-1995: Professor, Institute for Physical Science & Technology, and Department of Chemistry and Biochemistry, University of Maryland
- 1985-1990: Distinguished Member of Technical Staff, AT&T Bell Laboratories
- 1972-1985: Member of Technical Staff, AT&T Bell Laboratories
Research in the Weeks Group
Theoretical studies involving: nonuniform and confined fluids, ionic fluids, hydrophobic interactions, the static and dynamic properties of solid interfaces and thin films, density functional theory, pattern formation and crystal growth.
Major Recognitions and Honors
- Fellow, American Association for the Advancement of Science, 2011
- Member, National Academy of Sciences, USA 2009
- Colloquium Ehrenfestii, University of Leiden 2001
- Fellow, American Academy of Arts and Sciences, 2000
- Joel Henry Hildebrand Award in the Theoretical and Experimental Chemistry of Liquids (awarded by the American Chemical Society), 1990
- Regent’s Lecturer, University of California, Berkeley, 1990
- Fellow: American Physical Society, 1984
Significant Professional Service and Activities
1. Conferences and Society Organization:
- Chair, Subdivision of Theoretical Chemistry, American Chemical Society, 1992-1993
- Chair, Gordon Conference on Physics and Chemistry of Liquids, 1995; Vice-chair, 1993
2. Editorial Boards:
- Chemical Physics; Advances in Chemical Physics; Journal of Chemical Physics 1994 -1997
- Associate Editor, Journal of Statistical Physics 2011-
- Editorial Board, Proceedings of the National Academy of Sciences, 2012-
Research in the Weeks Group
We use fundamental methods of statistical mechanics to try to develop general quantitative theories that make contact with experiment. While we use computer simulations to refine and test our ideas, the main emphasis is on the development of new conceptual approaches where physical insight and intuition play an important role when properly coupled to statistical mechanics theory.
Charged and polar liquids present difficult problems on multiple length scales both for theory and in practical calculations. The long ranged nature of the Coulomb interactions generates long wavelength constraints like neutrality and dielectric screening phenomena that all equilibrium systems must obey, independent of the detailed nature of the short ranged intermolecular interactions. At the same time strong short ranged forces generate harshly repulsive molecular cores of various sizes and shapes that present specific packing constraints in the dense and disordered environment of a liquid near the triple point. Understanding and quantifying the interplay between long and short ranged forces in both simple and charged liquids has been a major focus of research in the Weeks group in recent years.
We have developed a new, physically motivated, and generally applicable approach called local molecular field (LMF) theory, by which one can determine equilibrium structure, thermodynamics, and phase transitions in nonuniform liquids with long-ranged Coulomb (and sometimes van der Waals (dispersion) interactions.) These interactions can be usefully separated into appropriately chosen long-ranged and slowly varying “far field” components and strong short ranged “near field” components.
LMF theory exploits in a controlled way the slowly varying nature of the far field components to provide a simplified but exceptionally accurate treatment of their effect on equilibrium structure and thermodynamics. These simplifications are particularly useful for systems (ions and water, in particular) with Coulomb interactions, whose long range presents problems both in molecular simulations and in theoretical attempts to provide a simple local picture of molecular correlations and dielectric screening. Two examples are highlighted below.
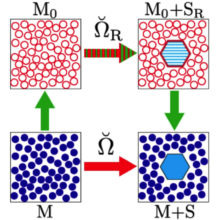 In R.C. Remsing, S. Liu, and J.D. Weeks, “Long-ranged contributions to solvation free energies from theory and short-ranged models” PNAS 113, 2819–2826 (2016) we show that LMF theory allows us to determine very accurately the long-ranged contributions to solvation free energies in charged and polar systems from models with only short-ranged, local interactions. We establish its accuracy for a variety of common solvation processes, including hydrophobic and ionic hydration, as well as colloidal overcharging. The figure illustrated the basic thermodynamic cycle we use to allow this simplification. The bottom leg illustrates solvation of a complex solute in the full solvent with complicated long ranged intermolecular interactions while the top illustrates solvation of a renormalized solute in a simplified solvent with only short ranged intermolecular interactions. LMF theory gives simple analytical expressions for the difference in solvation free energies, given by the sum of the green arrows.
In R.C. Remsing, S. Liu, and J.D. Weeks, “Long-ranged contributions to solvation free energies from theory and short-ranged models” PNAS 113, 2819–2826 (2016) we show that LMF theory allows us to determine very accurately the long-ranged contributions to solvation free energies in charged and polar systems from models with only short-ranged, local interactions. We establish its accuracy for a variety of common solvation processes, including hydrophobic and ionic hydration, as well as colloidal overcharging. The figure illustrated the basic thermodynamic cycle we use to allow this simplification. The bottom leg illustrates solvation of a complex solute in the full solvent with complicated long ranged intermolecular interactions while the top illustrates solvation of a renormalized solute in a simplified solvent with only short ranged intermolecular interactions. LMF theory gives simple analytical expressions for the difference in solvation free energies, given by the sum of the green arrows.
These ideas have recently been generalized to fluid mixtures and to the potential of mean force between a pair of charged mobile 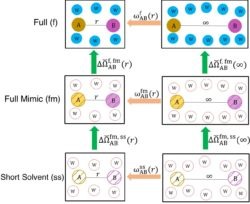 solutes in aqueous solution in A. Gao, R.C. Remsing, and J. D. Weeks, “Short solvent model for ion correlations and hydrophobic association. PNAS 117, 1293–1302 (2020). The resulting “short solvent” (SS) model has truncated solvent interactions and renormalized solute interactions and accurately describes the complex interplay between strong short-ranged solute core interactions, local hydrogen-bond configurations, and long-ranged dielectric screening of distant charges, competing effects that are very difficult to capture in standard implicit solvent models.
solutes in aqueous solution in A. Gao, R.C. Remsing, and J. D. Weeks, “Short solvent model for ion correlations and hydrophobic association. PNAS 117, 1293–1302 (2020). The resulting “short solvent” (SS) model has truncated solvent interactions and renormalized solute interactions and accurately describes the complex interplay between strong short-ranged solute core interactions, local hydrogen-bond configurations, and long-ranged dielectric screening of distant charges, competing effects that are very difficult to capture in standard implicit solvent models.
This figure illustrates the basic thermodynamic cycles that permit a very accurate determination of the pair correlation function between two ionic solutes in full water with untruncated Coulomb interactions (top panel) while requiring simulation results only from the much simpler SS model depicted in the bottom panel. Simple analytical expressions are derived for the green arrows connecting the various cycles.